- DIÉLECTRIQUES
- DIÉLECTRIQUESLe mot «diélectrique» s’emploie en général comme adjectif, pour qualifier une substance qui peut emmagasiner de l’énergie électrostatique, c’est-à-dire une substance à laquelle on peut appliquer un champ électrique élevé et sur laquelle on peut maintenir ce champ sans apport d’énergie extérieure. Par voie de conséquence, un matériau diélectrique doit être isolant.Toutefois, on parle souvent des propriétés diélectriques de matériaux non isolants (électrolytes, semi-conducteurs, métaux...). Il s’agit alors des propriétés liées au déphasage observé entre le champ et l’induction (ou déplacement) électriques, qui résulte du déplacement de charges et d’associations de charges (résonances, relaxations...) dans un champ sinusoïdal de faible amplitude.Ce sont essentiellement ces propriétés qui font l’objet du présent article, à l’exclusion de toutes les propriétés des isolants soumis à des champs intenses.Les impératifs de la miniaturisation ont réduit dans un rapport considérable le volume des éléments actifs d’un circuit, de sorte que les condensateurs, essentiellement passifs, prennent au moins autant de place que les éléments actifs. Les spécifications d’emploi ont également évolué. On cherche à utiliser des matériaux diélectriques à des températures variant entre quelques degrés absolus dans les circuits et moteurs cryotechniques, à plusieurs milliers de degrés dans les fours, les tuyères de réacteurs, les torches à plasma. Ils sont soumis à des conditions très rigoureuses (vapeurs d’eau, d’acides), à des sollicitations mécaniques statiques et dynamiques considérables, à des flux de radiation intenses au cœur des réacteurs nucléaires. On cherche à rester maître des charges statiques, qui, lors de décharges accidentelles, peuvent provoquer des catastrophes.De plus, les isolants et les diélectriques ont cessé de jouer le rôle passif d’obstacles au passage du courant et de réservoirs d’énergie électrique. Des diélectriques en couches minces permettent d’appliquer un champ électrique énorme à la surface d’un semi-conducteur, pour en moduler la conductivité. L’idée lancée par William B. Shockley, au début des années cinquante, d’un transistor à effet de champ, est largement exploitée commercialement.L’étude de l’injection d’un courant dans un diélectrique, sa limitation par charge d’espace et ses conséquences possibles, telles que la rectification et la résistance négative, est activement poursuivie. Il en est de même de l’émission contrôlée, par effet de champ, d’électrons «chauds» à travers une couche diélectrique très mince. Ces phénomènes pourraient déboucher dans un proche avenir sur de nouveaux composants électroniques.Par ailleurs, les diélectriques non linéaires , tels que les ferroélectriques, les antiferroélectriques, les piézoélectriques, ont des applications de plus en plus nombreuses: mémoires et bascules électroniques, obturation ultrarapide par effet Kerr ou effet Pockels, génération et détection des vibrations mécaniques, multiplication des fréquences optiques des faisceaux de laser.Parallèlement, les progrès de l’électronique dite classique ont amélioré considérablement les techniques d’étude des diélectriques. On sait mesurer des courants continus de 10-17 ampère, extraire un signal d’un bruit dans lequel il est noyé, analyser des phénomènes transitoires sous haute impédance, mesurer la permittivité des corps dans une très large gamme de fréquences (de 10-3 à 1011 Hz), mesurer des facteurs de dissipation très faibles, de l’ordre de 10-5, ce qui, dans la recherche, confère aux diélectriques une place à peu près analogue à celle des semi-conducteurs en 1945.On ne traitera ici que des bases théoriques élémentaires permettant d’introduire les notions qui seront développées dans les articles se référant aux applications qu’on vient d’esquisser.1. Rappel d’électrostatique du videDans toute la suite du texte, on désigne par la lettre grecque 﨎 une permittivité absolue et par la lettre une permittivité relative par rapport au vide.La loi fondamentale de l’électrostatique, ou loi de Coulomb :
 exprime la force répulsive s’exerçant entre deux charges ponctuelles q situées dans le vide et séparées par la distance r . Cette relation (1) définit l’unité de charge électrostatique C.G.S. comme celle qui, placée à 1 centimètre d’une autre charge identique, est soumise à une force répulsive de 1 dyne.Dans le système S.I., généralement utilisé aujourd’hui, l’unité de charge est définie comme la charge transportée par un courant de 1 ampère pendant 1 seconde: 1 coulomb (C) = 1 A.s. Le coulomb et l’unité électrostatique C.G.S. sont liés par une relation où figure la vitesse c de propagation d’une onde électromagnétique dans le vide (cf. ÉLECTRICITÉ – Électromagnétisme). En prenant, ce qui est une approximation, c = 3.108 m s-1, on montre que le coulomb vaut 3.109 u.e.s. C.G.S., de sorte que la relation (1), exprimée dans le système S.I., devient:
exprime la force répulsive s’exerçant entre deux charges ponctuelles q situées dans le vide et séparées par la distance r . Cette relation (1) définit l’unité de charge électrostatique C.G.S. comme celle qui, placée à 1 centimètre d’une autre charge identique, est soumise à une force répulsive de 1 dyne.Dans le système S.I., généralement utilisé aujourd’hui, l’unité de charge est définie comme la charge transportée par un courant de 1 ampère pendant 1 seconde: 1 coulomb (C) = 1 A.s. Le coulomb et l’unité électrostatique C.G.S. sont liés par une relation où figure la vitesse c de propagation d’une onde électromagnétique dans le vide (cf. ÉLECTRICITÉ – Électromagnétisme). En prenant, ce qui est une approximation, c = 3.108 m s-1, on montre que le coulomb vaut 3.109 u.e.s. C.G.S., de sorte que la relation (1), exprimée dans le système S.I., devient: Les relations (1) ou (2) permettent de trouver le champ électrique E = F /q créé par une charge ponctuelle q dans le vide, à la distance r de cette charge. Ce champ s’exprime en statvolts par centimètre (système C.G.S.) ou en volts par mètre (système S.I.).L’induction, ou déplacement, électrique D peut être définie à partir de la loi de Gauss , d’après laquelle le flux d’induction à travers une surface gauche fermée est égal à la somme des charges contenues à l’intérieur de cette surface. Dans le vide, le vecteur induction っ est proportionnel au vecteur champ つ, et le coefficient de proportionnalité est une constante 﨎 0, caractéristique du vide, qu’on appelle permittivité du vide. En appliquant la loi de Gauss à une sphère de rayon r au centre de laquelle se trouve une charge q , on trouve:
Les relations (1) ou (2) permettent de trouver le champ électrique E = F /q créé par une charge ponctuelle q dans le vide, à la distance r de cette charge. Ce champ s’exprime en statvolts par centimètre (système C.G.S.) ou en volts par mètre (système S.I.).L’induction, ou déplacement, électrique D peut être définie à partir de la loi de Gauss , d’après laquelle le flux d’induction à travers une surface gauche fermée est égal à la somme des charges contenues à l’intérieur de cette surface. Dans le vide, le vecteur induction っ est proportionnel au vecteur champ つ, et le coefficient de proportionnalité est une constante 﨎 0, caractéristique du vide, qu’on appelle permittivité du vide. En appliquant la loi de Gauss à une sphère de rayon r au centre de laquelle se trouve une charge q , on trouve: soit:
soit: En identifiant cette expression du champ à celle qui est obtenue en divisant F par q dans la formule (2), on obtient la valeur numérique de la constante 﨎 0:
En identifiant cette expression du champ à celle qui est obtenue en divisant F par q dans la formule (2), on obtient la valeur numérique de la constante 﨎 0: On peut également définir 﨎 0 à l’aide d’un condensateur plan à anneau de garde (pour assurer l’uniformité du champ entre les armatures considérées). Les armatures actives, de surface S , sont séparées par l’intervalle d (fig. 1).Si l’on applique une différence de potentiel V entre ces armatures, celles-ci prennent des charges Q et 漣 Q uniformément réparties, et le champ électrique entre les armatures est uniforme:
On peut également définir 﨎 0 à l’aide d’un condensateur plan à anneau de garde (pour assurer l’uniformité du champ entre les armatures considérées). Les armatures actives, de surface S , sont séparées par l’intervalle d (fig. 1).Si l’on applique une différence de potentiel V entre ces armatures, celles-ci prennent des charges Q et 漣 Q uniformément réparties, et le champ électrique entre les armatures est uniforme: Soit 梁 靖 = 梁 Q /S la densité superficielle de charges sur les armatures. L’application de la loi de Gauss au petit cylindre représenté sur la figure donne:
Soit 梁 靖 = 梁 Q /S la densité superficielle de charges sur les armatures. L’application de la loi de Gauss au petit cylindre représenté sur la figure donne: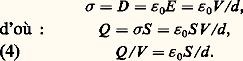 Le second membre de (4) est indépendant de la tension appliquée, ce qui montre que Q est proportionnel à V . Le rapport Q /V (coulombs par volt), qui ne dépend que des dimensions du condensateur, est sa capacité à vide , exprimée en farads (F), et l’équation (4) montre que la constante 﨎 0 s’exprime en farads par mètre.Supposons qu’on applique entre les armatures d’un condensateur à vide une tension sinusoïdale de la forme:
Le second membre de (4) est indépendant de la tension appliquée, ce qui montre que Q est proportionnel à V . Le rapport Q /V (coulombs par volt), qui ne dépend que des dimensions du condensateur, est sa capacité à vide , exprimée en farads (F), et l’équation (4) montre que la constante 﨎 0 s’exprime en farads par mètre.Supposons qu’on applique entre les armatures d’un condensateur à vide une tension sinusoïdale de la forme: la charge Q accumulée sur les armatures du condensateur est en phase avec la tension V , et vaut Q = C 0V avec, en vertu de (4), C 0 = 﨎 0S /d .En revanche, le courant I qui passe dans le circuit extérieur, égal à la dérivée de la charge Q par rapport au temps:
la charge Q accumulée sur les armatures du condensateur est en phase avec la tension V , et vaut Q = C 0V avec, en vertu de (4), C 0 = 﨎 0S /d .En revanche, le courant I qui passe dans le circuit extérieur, égal à la dérivée de la charge Q par rapport au temps: est en quadrature de phase par rapport à la tension appliquée V . C’est un courant «de déplacement» non dissipatif .2. Permittivité d’un diélectrique isotropeCondensateur à diélectriqueRemplissons le condensateur avec un fluide diélectrique. La valeur C de la capacité du condensateur ainsi rempli est supérieure à C 0, et le rapport C /C 0 est la permittivité relative du matériau. Le «prime» qui est affecté au symbole indique qu’il s’agit d’une quantité réelle. On verra plus loin que la dissipation fait apparaître une composante imaginaire de la permittivité, qu’il faut distinguer de sa composante réelle. Chaque fois que la permittivité est réelle, le «prime» pourra être supprimé sans ambiguïté.Pour comprendre l’origine physique de l’augmentation de la capacité qui résulte de l’introduction d’un fluide diélectrique dans le condensateur, on peut faire le raisonnement suivant: chargeons le condensateur vide à la tension V ; par définition de C 0, des charges 梁 Q = 梁 C 0V apparaissent sur les électrodes; déconnectons maintenant le condensateur de la source de tension et introduisons le fluide en évitant de décharger le condensateur; la charge Q reste constante, mais une partie de cette charge, maintenant immobilisée par des dipôles (induits ou permanents), ne contribue plus à maintenir la différence de potentiel entre les armatures; cette différence de potentiel diminue, mais cela n’est possible que si la capacité C a augmenté, puisque le produit CV = Q est constant. Notons que, si on avait rempli le condensateur sous tension constante, l’augmentation de capacité aurait produit un courant dans le circuit extérieur.La permittivité relative des matériaux diélectriques aux fréquences usuelles est toujours supérieure à l’unité, valeur qui correspond au vide. Elle varie avec la concentration et la nature des dipôles. Pour un matériau condensé non polaire , c’est-à-dire ne contenant pas de dipôles permanents (gaz élémentaires liquéfiés, hydrocarbures simples, etc.), les seuls dipôles contribuant à sont des dipôles induits par la polarisation des atomes ou des molécules dans le champ électrique, comme on le verra plus loin, et la valeur de est de l’ordre de 2 (de 1,5 à 4 environ). En revanche, pour un matériau polaire, c’est-à-dire constitué de dipôles (H2O, C2, etc.), la valeur de aux fréquences assez basses (cf. chap. 10) peut dépasser 100; quant aux matériaux ferroélectriques, leur permittivité relative juste au-dessus du point de Curie peut atteindre plusieurs milliers.La relation っ = 﨎 0 つ qui relie dans le vide l’induction っ au champ つ doit être remplacée par っ = 﨎 つ = 﨎 0 つ = 﨎 0 つ + は, où は est le vecteur polarisation:
est en quadrature de phase par rapport à la tension appliquée V . C’est un courant «de déplacement» non dissipatif .2. Permittivité d’un diélectrique isotropeCondensateur à diélectriqueRemplissons le condensateur avec un fluide diélectrique. La valeur C de la capacité du condensateur ainsi rempli est supérieure à C 0, et le rapport C /C 0 est la permittivité relative du matériau. Le «prime» qui est affecté au symbole indique qu’il s’agit d’une quantité réelle. On verra plus loin que la dissipation fait apparaître une composante imaginaire de la permittivité, qu’il faut distinguer de sa composante réelle. Chaque fois que la permittivité est réelle, le «prime» pourra être supprimé sans ambiguïté.Pour comprendre l’origine physique de l’augmentation de la capacité qui résulte de l’introduction d’un fluide diélectrique dans le condensateur, on peut faire le raisonnement suivant: chargeons le condensateur vide à la tension V ; par définition de C 0, des charges 梁 Q = 梁 C 0V apparaissent sur les électrodes; déconnectons maintenant le condensateur de la source de tension et introduisons le fluide en évitant de décharger le condensateur; la charge Q reste constante, mais une partie de cette charge, maintenant immobilisée par des dipôles (induits ou permanents), ne contribue plus à maintenir la différence de potentiel entre les armatures; cette différence de potentiel diminue, mais cela n’est possible que si la capacité C a augmenté, puisque le produit CV = Q est constant. Notons que, si on avait rempli le condensateur sous tension constante, l’augmentation de capacité aurait produit un courant dans le circuit extérieur.La permittivité relative des matériaux diélectriques aux fréquences usuelles est toujours supérieure à l’unité, valeur qui correspond au vide. Elle varie avec la concentration et la nature des dipôles. Pour un matériau condensé non polaire , c’est-à-dire ne contenant pas de dipôles permanents (gaz élémentaires liquéfiés, hydrocarbures simples, etc.), les seuls dipôles contribuant à sont des dipôles induits par la polarisation des atomes ou des molécules dans le champ électrique, comme on le verra plus loin, et la valeur de est de l’ordre de 2 (de 1,5 à 4 environ). En revanche, pour un matériau polaire, c’est-à-dire constitué de dipôles (H2O, C2, etc.), la valeur de aux fréquences assez basses (cf. chap. 10) peut dépasser 100; quant aux matériaux ferroélectriques, leur permittivité relative juste au-dessus du point de Curie peut atteindre plusieurs milliers.La relation っ = 﨎 0 つ qui relie dans le vide l’induction っ au champ つ doit être remplacée par っ = 﨎 つ = 﨎 0 つ = 﨎 0 つ + は, où は est le vecteur polarisation: Composante dissipative de la permittivitéSimultanément avec l’accroissement de capacité, on observe en général une composante du courant alternatif en phase avec la tension appliquée.Cette composante I p = GV , où G est la conductance de l’échantillon (G = R -1 = 靖 Sd -1), représente un courant dissipatif, en phase avec V .Le courant total est, dans ce cas, la somme vectorielle:
Composante dissipative de la permittivitéSimultanément avec l’accroissement de capacité, on observe en général une composante du courant alternatif en phase avec la tension appliquée.Cette composante I p = GV , où G est la conductance de l’échantillon (G = R -1 = 靖 Sd -1), représente un courant dissipatif, en phase avec V .Le courant total est, dans ce cas, la somme vectorielle: ou, en revenant aux caractéristiques du matériau:
ou, en revenant aux caractéristiques du matériau: Conductivité complexe size=5靖La relation (8) peut s’écrire sous la forme
Conductivité complexe size=5靖La relation (8) peut s’écrire sous la forme avec 靖 ; = 靖 + j 諸 﨎 .Le déphasage entre I et V , qui était exactement égal à 神/2 avant le remplissage du condensateur, diminue d’un petit angle 嗀 , appelé angle de pertes (fig. 2), défini par:
avec 靖 ; = 靖 + j 諸 﨎 .Le déphasage entre I et V , qui était exactement égal à 神/2 avant le remplissage du condensateur, diminue d’un petit angle 嗀 , appelé angle de pertes (fig. 2), défini par: En pratique, si les caractéristiques connues du matériau sont sa résistivité 福 , sa permittivité relative et si la fréquence f est exprimée en hertz, la relation (10) devient:
En pratique, si les caractéristiques connues du matériau sont sa résistivité 福 , sa permittivité relative et si la fréquence f est exprimée en hertz, la relation (10) devient: Le tableau 1 se réfère à un cas typique.Circuit équivalentLe raisonnement que l’on vient de faire nous a amené à caractériser un matériau diélectrique par sa permittivité et sa conductivité. En supposant que ces deux quantités sont réellement des constantes, on est donc amené à représenter le matériau par un circuit équivalent RC , le bras capacitif correspondant à la permittivité et le bras résistif à la résistivité (ou conductivité). Un tel circuit prévoit une variation de tan 嗀 proportionnelle à l’inverse de la fréquence (fig. 3).La déviation généralement observée par rapport à cette loi témoigne du fait que la permittivité du matériau, loin d’être constante, n’est définie que dans des conditions particulières de fréquence et de température. Il faut donc éviter de parler de «constante diélectrique» pour un matériau autre que le vide.3. Permittivité complexePermittivité complexe absolue size=5﨎La relation (8) peut s’écrire sous la forme:
Le tableau 1 se réfère à un cas typique.Circuit équivalentLe raisonnement que l’on vient de faire nous a amené à caractériser un matériau diélectrique par sa permittivité et sa conductivité. En supposant que ces deux quantités sont réellement des constantes, on est donc amené à représenter le matériau par un circuit équivalent RC , le bras capacitif correspondant à la permittivité et le bras résistif à la résistivité (ou conductivité). Un tel circuit prévoit une variation de tan 嗀 proportionnelle à l’inverse de la fréquence (fig. 3).La déviation généralement observée par rapport à cette loi témoigne du fait que la permittivité du matériau, loin d’être constante, n’est définie que dans des conditions particulières de fréquence et de température. Il faut donc éviter de parler de «constante diélectrique» pour un matériau autre que le vide.3. Permittivité complexePermittivité complexe absolue size=5﨎La relation (8) peut s’écrire sous la forme:
 Compte tenu de (10), la relation (13) devient:
Compte tenu de (10), la relation (13) devient: La relation (13) montre que si la conductivité 靖 due par exemple à des porteurs libres, est une constante du matériau, la composante imaginaire 﨎 de 﨎 varie comme l’inverse de la fréquence. La relation (14) montre que si au contraire le matériau peut être caractérisé par son angle de pertes 嗀 , la conductivité équivalente est proportionnelle à la fréquence.Propagation d’une onde dans un diélectriqueIl résulte des équations de Maxwell (cf. ÉLECTRICITÉ – Électromagnétisme) qu’une onde électromagnétique du type:
La relation (13) montre que si la conductivité 靖 due par exemple à des porteurs libres, est une constante du matériau, la composante imaginaire 﨎 de 﨎 varie comme l’inverse de la fréquence. La relation (14) montre que si au contraire le matériau peut être caractérisé par son angle de pertes 嗀 , la conductivité équivalente est proportionnelle à la fréquence.Propagation d’une onde dans un diélectriqueIl résulte des équations de Maxwell (cf. ÉLECTRICITÉ – Électromagnétisme) qu’une onde électromagnétique du type: où 諸 est la pulsation et 塚 le coefficient d’amortissement complexe ( 塚 = 見 + j 廓 ), se propage dans un matériau non magnétique, de perméabilité 猪 voisine de celle du vide 猪 0, à la vitesse 羽 = ( 猪 0 﨎 ) 漣1/2.Dans le vide, c = ( 猪 0 﨎 0) 漣1/2, de sorte que l’indice de réfraction complexe face="EU Domacr" 滑 pour l’onde considérée dans le diélectrique est:
où 諸 est la pulsation et 塚 le coefficient d’amortissement complexe ( 塚 = 見 + j 廓 ), se propage dans un matériau non magnétique, de perméabilité 猪 voisine de celle du vide 猪 0, à la vitesse 羽 = ( 猪 0 﨎 ) 漣1/2.Dans le vide, c = ( 猪 0 﨎 0) 漣1/2, de sorte que l’indice de réfraction complexe face="EU Domacr" 滑 pour l’onde considérée dans le diélectrique est: k étant l’indice d’absorption.L’identification des parties réelles et imaginaires dans la relation ci-dessus se réduit, en supposant 廉 1 à:
k étant l’indice d’absorption.L’identification des parties réelles et imaginaires dans la relation ci-dessus se réduit, en supposant 廉 1 à: Le coefficient d’amortissement complexe 塚 est lié à face="EU Domacr" 滑 par 塚 = (j 諸 /c ) face="EU Domacr" 滑 , d’où:
Le coefficient d’amortissement complexe 塚 est lié à face="EU Domacr" 滑 par 塚 = (j 諸 /c ) face="EU Domacr" 滑 , d’où: La mesure du coefficient d’amortissement 見 et du coefficient de phase 廓 permet de connaître k et n , donc et . Cela constitue la base, simplifiée il est vrai, de la mesure des permittivités en hyperfréquences.Nature tensorielle de la permittivitéDans le cas particulier d’un matériau isotrope et linéaire (c’est-à-dire où | っ| est proportionnel à | つ|), l’induction est colinéaire et proportionnelle au champ électrique. Dans ce cas, la permittivité est un scalaire , et le matériau est dit de classe A.Dans un matériau non isotrope (un cristal par exemple), l’induction et le champ ne sont pas colinéaires en général, et la permittivité est un tenseur . Toutefois, on montre facilement que dans le cas d’un cristal à symétrie sphérique (cristal cubique), la permittivité est un scalaire dans la mesure où le champ est faible.4. Énergétique des diélectriquesPour augmenter de dw l’énergie libre électrique d’un volume unitaire de diélectrique soumis au champ つ, il faut fournir, contre les forces électrostatiques, le travail élémentaire つ.d っ. On peut en déduire que la densité d’énergie dans un cristal est w = つ. っ/2.Pour un matériau de classe A, cette dernière relation se réduit à:
La mesure du coefficient d’amortissement 見 et du coefficient de phase 廓 permet de connaître k et n , donc et . Cela constitue la base, simplifiée il est vrai, de la mesure des permittivités en hyperfréquences.Nature tensorielle de la permittivitéDans le cas particulier d’un matériau isotrope et linéaire (c’est-à-dire où | っ| est proportionnel à | つ|), l’induction est colinéaire et proportionnelle au champ électrique. Dans ce cas, la permittivité est un scalaire , et le matériau est dit de classe A.Dans un matériau non isotrope (un cristal par exemple), l’induction et le champ ne sont pas colinéaires en général, et la permittivité est un tenseur . Toutefois, on montre facilement que dans le cas d’un cristal à symétrie sphérique (cristal cubique), la permittivité est un scalaire dans la mesure où le champ est faible.4. Énergétique des diélectriquesPour augmenter de dw l’énergie libre électrique d’un volume unitaire de diélectrique soumis au champ つ, il faut fournir, contre les forces électrostatiques, le travail élémentaire つ.d っ. On peut en déduire que la densité d’énergie dans un cristal est w = つ. っ/2.Pour un matériau de classe A, cette dernière relation se réduit à: Pour emmagasiner autant d’énergie que possible par unité de volume dans l’isolement d’un condensateur, il faut choisir un matériau tel que le produit 﨎 E 2 de la permittivité par la rigidité diélectrique soit maximal. Le maximum théorique de w est de l’ordre de 2.105 J.m-3. Compte tenu des armatures, enveloppes et conditionnement, la densité d’énergie emmagasinée dans les condensateurs dépasse rarement 103 J.m-3. Il reste encore à faire de grands progrès dans ce domaine.Effet électrocaloriquePour établir la relation (16) on ne tient pas compte du fait qu’en chargeant un diélectrique polaire de E = 0 à E , on tend à le réchauffer, par un processus thermodynamique réversible , tout à fait différent d’une dissipation qui, elle, est irréversible. En effet, à l’orientation des dipôles, provoquée par le champ, correspond un abaissement de l’entropie du système de ces dipôles. Les dipôles vont donc céder de l’énergie thermique à leur environnement, dont la température tend à augmenter. Inversement, l’annulation du champ va tendre à refroidir l’environnement. Par suite, le travail nécessaire pour charger le diélectrique de façon isotherme n’est plus exactement donné par (16), mais par:
Pour emmagasiner autant d’énergie que possible par unité de volume dans l’isolement d’un condensateur, il faut choisir un matériau tel que le produit 﨎 E 2 de la permittivité par la rigidité diélectrique soit maximal. Le maximum théorique de w est de l’ordre de 2.105 J.m-3. Compte tenu des armatures, enveloppes et conditionnement, la densité d’énergie emmagasinée dans les condensateurs dépasse rarement 103 J.m-3. Il reste encore à faire de grands progrès dans ce domaine.Effet électrocaloriquePour établir la relation (16) on ne tient pas compte du fait qu’en chargeant un diélectrique polaire de E = 0 à E , on tend à le réchauffer, par un processus thermodynamique réversible , tout à fait différent d’une dissipation qui, elle, est irréversible. En effet, à l’orientation des dipôles, provoquée par le champ, correspond un abaissement de l’entropie du système de ces dipôles. Les dipôles vont donc céder de l’énergie thermique à leur environnement, dont la température tend à augmenter. Inversement, l’annulation du champ va tendre à refroidir l’environnement. Par suite, le travail nécessaire pour charger le diélectrique de façon isotherme n’est plus exactement donné par (16), mais par: Il résulte de la loi de Langevin que la composante de 﨎 due à l’orientation des dipôles 猪 (en concentration N ) est:
Il résulte de la loi de Langevin que la composante de 﨎 due à l’orientation des dipôles 猪 (en concentration N ) est: et par suite:
et par suite: Le terme correctif de (17) correspond à la quantité de chaleur qu’il faut enlever au matériau ( 煉 﨎 / 煉 T 麗 0) pour le maintenir isotherme, et ce terme est d’autant plus grand que la température est basse.Le refroidissement d’un système de dipôles par effet électrocalorique a été observé en 1964, et cet effet semble pouvoir être utilisé, parallèlement à la désaimantation adiabatique, pour l’obtention de très basses températures.Le second terme de (17) peut également devenir important pour un matériau ferroélectrique au voisinage du point de transition, et des applications à la conversion de l’énergie ont été proposées.5. Champ dépolarisant et facteur de dépolarisationSoumettons un matériau diélectrique initialement neutre à un champ extérieur. Ce champ induit des dipôles dans le matériau. Ces dipôles induits produisent eux-mêmes des champs dont la résultante tend à s’opposer au champ extérieur.Le champ dépolarisant est le champ E dép de réaction créé par les dipôles induits; c’est la différence entre le champ initial extérieur et le champ interne dans le matériau après établissement de la polarisation.Le facteur dépolarisant est par définition le rapport 塚 = 﨎 0E dép/P. Dans la plupart des cas (diélectriques linéaires), c’est un facteur numérique ne dépendant que de la forme de l’échantillon et de la direction du champ appliqué. Sa connaissance permet de calculer le moment dipolaire pris par un petit échantillon diélectrique dans un champ.Examinons quelques exemples.Considérons tout d’abord le problème à une dimension de la plaque mince d’un matériau non chargé de permittivité relative , perpendiculaire au champ extérieur (la plaque est supposée dans le vide). La polarisation et le champ à l’intérieur sont uniformes, et il n’y a pas de charge à l’intérieur. L’effet des dipôles se réduit à la présence de deux couches de charges superficielles (liées) de signes opposés, dont l’épaisseur est comparable aux dimensions atomiques, qui créent le champ dépolarisant.Comme il n’y a pas de charge superficielle libre, D doit être continu à l’interface, ce qui entraîne:
Le terme correctif de (17) correspond à la quantité de chaleur qu’il faut enlever au matériau ( 煉 﨎 / 煉 T 麗 0) pour le maintenir isotherme, et ce terme est d’autant plus grand que la température est basse.Le refroidissement d’un système de dipôles par effet électrocalorique a été observé en 1964, et cet effet semble pouvoir être utilisé, parallèlement à la désaimantation adiabatique, pour l’obtention de très basses températures.Le second terme de (17) peut également devenir important pour un matériau ferroélectrique au voisinage du point de transition, et des applications à la conversion de l’énergie ont été proposées.5. Champ dépolarisant et facteur de dépolarisationSoumettons un matériau diélectrique initialement neutre à un champ extérieur. Ce champ induit des dipôles dans le matériau. Ces dipôles induits produisent eux-mêmes des champs dont la résultante tend à s’opposer au champ extérieur.Le champ dépolarisant est le champ E dép de réaction créé par les dipôles induits; c’est la différence entre le champ initial extérieur et le champ interne dans le matériau après établissement de la polarisation.Le facteur dépolarisant est par définition le rapport 塚 = 﨎 0E dép/P. Dans la plupart des cas (diélectriques linéaires), c’est un facteur numérique ne dépendant que de la forme de l’échantillon et de la direction du champ appliqué. Sa connaissance permet de calculer le moment dipolaire pris par un petit échantillon diélectrique dans un champ.Examinons quelques exemples.Considérons tout d’abord le problème à une dimension de la plaque mince d’un matériau non chargé de permittivité relative , perpendiculaire au champ extérieur (la plaque est supposée dans le vide). La polarisation et le champ à l’intérieur sont uniformes, et il n’y a pas de charge à l’intérieur. L’effet des dipôles se réduit à la présence de deux couches de charges superficielles (liées) de signes opposés, dont l’épaisseur est comparable aux dimensions atomiques, qui créent le champ dépolarisant.Comme il n’y a pas de charge superficielle libre, D doit être continu à l’interface, ce qui entraîne:
 et le facteur dépolarisant 塚 est égal à l’unité.Le second exemple est celui de la sphère diélectrique de rayon R et de permittivité relative dans un champ extérieur (la sphère est supposée dans le vide).En tout point de l’espace, le potentiel V obéit à l’équation de Laplace 暴2V = 0. La solution générale de cette équation, présentant une symétrie de révolution autour de la direction OZ du champ extérieur, est:
et le facteur dépolarisant 塚 est égal à l’unité.Le second exemple est celui de la sphère diélectrique de rayon R et de permittivité relative dans un champ extérieur (la sphère est supposée dans le vide).En tout point de l’espace, le potentiel V obéit à l’équation de Laplace 暴2V = 0. La solution générale de cette équation, présentant une symétrie de révolution autour de la direction OZ du champ extérieur, est: où A n et B n sont des constantes qui vont être déterminées par les conditions aux limites et où P n (cos ) désigne le polynôme de Legendre d’ordre n en cos . Il existe une fonction potentiel du type (19) pour l’intérieur (V i) et une autre fonction potentiel du même type pour l’extérieur (V e). V e doit être tel que, loin de la sphère, on retrouve le potentiel non perturbé, ce qui implique que tous les B en sont nuls sauf B e1, qui doit être égal à ( face=F0019 漣 E 0).En prenant l’origine au centre de la sphère, on voit que tous les A in doivent être nuls. Finalement, on doit écrire que le potentiel V et l’induction ( 﨎 煉 V / 煉 r ) sont tous deux continus à l’interface (r = R ). Cela n’est possible que si tous les coefficients d’indice n différent de 1 sont nuls.On en déduit:
où A n et B n sont des constantes qui vont être déterminées par les conditions aux limites et où P n (cos ) désigne le polynôme de Legendre d’ordre n en cos . Il existe une fonction potentiel du type (19) pour l’intérieur (V i) et une autre fonction potentiel du même type pour l’extérieur (V e). V e doit être tel que, loin de la sphère, on retrouve le potentiel non perturbé, ce qui implique que tous les B en sont nuls sauf B e1, qui doit être égal à ( face=F0019 漣 E 0).En prenant l’origine au centre de la sphère, on voit que tous les A in doivent être nuls. Finalement, on doit écrire que le potentiel V et l’induction ( 﨎 煉 V / 煉 r ) sont tous deux continus à l’interface (r = R ). Cela n’est possible que si tous les coefficients d’indice n différent de 1 sont nuls.On en déduit: ce qui montre que le champ à l’intérieur de la sphère est uniforme:
ce qui montre que le champ à l’intérieur de la sphère est uniforme: Il en est de même pour le champ dépolarisant:
Il en est de même pour le champ dépolarisant: d’où 塚 = 1/3.Le champ et l’induction sont uniformes à l’intérieur de la sphère. Pour = 10, par exemple:
d’où 塚 = 1/3.Le champ et l’induction sont uniformes à l’intérieur de la sphère. Pour = 10, par exemple: d’où l’allure des courbes de la figure 4, sur laquelle les valeurs du champ つ et de l’induction っ à l’intérieur et à l’extérieur sont représentées par un trait plus ou moins gros. Ces résultats ont été généralisés au cas d’un ellipsoïde. Un résumé des résultats relatifs aux configurations les plus simples est donné dans la figure 5.6. Champ local et relation de Clausius-MossottiJusqu’ici, on a considéré la polarisation sous son aspect macroscopique, et on a vu que, dans un diélectrique isotrope et linéaire (c’est-à-dire de classe A), face="EU Arrow" は s’exprime en fonction du champ つ par la relation は = 﨎 0 﨑 つ, 﨑 étant la susceptibilité électrique scalaire.À l’échelle moléculaire, cette polarisation résulte d’un déplacement relatif des charges négatives (électrons et anions) par rapport aux charges positives (noyaux et cations) et c’est ce déplacement relatif qui se manifeste par la charge électrique superficielle mentionnée plus haut.Cas des diélectriques diluésPar définition, la polarisabilité 見 d’une molécule est le moment dipolaire qu’elle acquiert dans un champ électrique unité. Dans un champ つ , le moment dipolaire de la molécule polarisée est 見 つ . Si N est la concentration des molécules, et si l’on suppose que ces molécules sont assez dispersées pour que la présence des dipôles induits ne modifie pas sensiblement la valeur du champ, つ = つ et on peut relier la grandeur macroscopique 﨑 à la grandeur microscopique 見 , en exprimant de deux façons différentes la polarisation P :
d’où l’allure des courbes de la figure 4, sur laquelle les valeurs du champ つ et de l’induction っ à l’intérieur et à l’extérieur sont représentées par un trait plus ou moins gros. Ces résultats ont été généralisés au cas d’un ellipsoïde. Un résumé des résultats relatifs aux configurations les plus simples est donné dans la figure 5.6. Champ local et relation de Clausius-MossottiJusqu’ici, on a considéré la polarisation sous son aspect macroscopique, et on a vu que, dans un diélectrique isotrope et linéaire (c’est-à-dire de classe A), face="EU Arrow" は s’exprime en fonction du champ つ par la relation は = 﨎 0 﨑 つ, 﨑 étant la susceptibilité électrique scalaire.À l’échelle moléculaire, cette polarisation résulte d’un déplacement relatif des charges négatives (électrons et anions) par rapport aux charges positives (noyaux et cations) et c’est ce déplacement relatif qui se manifeste par la charge électrique superficielle mentionnée plus haut.Cas des diélectriques diluésPar définition, la polarisabilité 見 d’une molécule est le moment dipolaire qu’elle acquiert dans un champ électrique unité. Dans un champ つ , le moment dipolaire de la molécule polarisée est 見 つ . Si N est la concentration des molécules, et si l’on suppose que ces molécules sont assez dispersées pour que la présence des dipôles induits ne modifie pas sensiblement la valeur du champ, つ = つ et on peut relier la grandeur macroscopique 﨑 à la grandeur microscopique 見 , en exprimant de deux façons différentes la polarisation P : Cas des diélectriques condensésSi, maintenant, on considère un milieu dans lequel les particules polaires ou polarisables sont beaucoup plus concentrées (gaz comprimés, liquides ou solides), la relation (20) n’est plus valable, car chaque molécule «voit» un champ différent de つ. En effet, en plus de つ, chaque molécule «voit» le champ dépolarisant dû aux distributions superficielles, ainsi que les champs de tous les dipôles voisins.Le calcul du champ électrique «vu» par un atome polarisable en un point A d’un diélectrique condensé est un des problèmes centraux de la théorie des diélectriques.Pour trouver le champ local en un point A d’une plaque diélectrique perpendiculaire au champ extérieur, on commence par calculer le champ interne macroscopique en soustrayant le champ dépolarisant du champ extérieur: face="EU Arrow" つ = つ0 漣 つdép.Le champ créé en A par un dipôle centré en ce point s’oppose à つ. Par suite, retirer ce dipôle du matériau revient à créer en A un champ supérieur à つ. Cela montre que le champ «local» en A est supérieur à つ. Pour calculer de combien le champ «local» dépasse つ, on extrait par la pensée une petite sphère de centre A en maintenant tous les dipôles extérieurs à la sphère dans la position moyenne qu’ils occupaient avant l’extraction de la sphère, pour éviter de modifier les lignes de force à l’extérieur.Il apparaît ainsi une charge superficielle correspondant aux extrémités des dipôles à la surface interne de la cavité fictive (de Mossotti), et le champ créé par cette charge superficielle en A est つs.On peut alors remettre les dipôles un à un dans la cavité, excepté le dipôle A au milieu duquel on cherche le champ, et enfin tenir compte du champ つcav créé par ces dipôles en A.On a donc:
Cas des diélectriques condensésSi, maintenant, on considère un milieu dans lequel les particules polaires ou polarisables sont beaucoup plus concentrées (gaz comprimés, liquides ou solides), la relation (20) n’est plus valable, car chaque molécule «voit» un champ différent de つ. En effet, en plus de つ, chaque molécule «voit» le champ dépolarisant dû aux distributions superficielles, ainsi que les champs de tous les dipôles voisins.Le calcul du champ électrique «vu» par un atome polarisable en un point A d’un diélectrique condensé est un des problèmes centraux de la théorie des diélectriques.Pour trouver le champ local en un point A d’une plaque diélectrique perpendiculaire au champ extérieur, on commence par calculer le champ interne macroscopique en soustrayant le champ dépolarisant du champ extérieur: face="EU Arrow" つ = つ0 漣 つdép.Le champ créé en A par un dipôle centré en ce point s’oppose à つ. Par suite, retirer ce dipôle du matériau revient à créer en A un champ supérieur à つ. Cela montre que le champ «local» en A est supérieur à つ. Pour calculer de combien le champ «local» dépasse つ, on extrait par la pensée une petite sphère de centre A en maintenant tous les dipôles extérieurs à la sphère dans la position moyenne qu’ils occupaient avant l’extraction de la sphère, pour éviter de modifier les lignes de force à l’extérieur.Il apparaît ainsi une charge superficielle correspondant aux extrémités des dipôles à la surface interne de la cavité fictive (de Mossotti), et le champ créé par cette charge superficielle en A est つs.On peut alors remettre les dipôles un à un dans la cavité, excepté le dipôle A au milieu duquel on cherche le champ, et enfin tenir compte du champ つcav créé par ces dipôles en A.On a donc: toutes ces composantes étant dirigées suivant つ0 à cause de la symétrie de la configuration.On évalue les trois dernières composantes:– le champ dépolarisant つdép = は/ 﨎 0;– le champ des charges superficielles つs; un calcul classique conduit à la relation つs = は/3 﨎 0;– le champ de cavité つcav (champ des dipôles dans la cavité); d’une façon générale, ce champ est nul dans les diélectriques de classe A. E cav est nul également dans les matériaux cristallins dont la maille a un centre de symétrie (halogénures alcalins par exemple). Bien que certaines propriétés physiques des cristaux cubiques ne soient pas isotropes (les propriétés de transport, de cohésion, d’absorption optique, etc., peuvent varier avec la direction cristalline), on peut montrer que le champ de cavité est nul dans ces cristaux, et par suite:
toutes ces composantes étant dirigées suivant つ0 à cause de la symétrie de la configuration.On évalue les trois dernières composantes:– le champ dépolarisant つdép = は/ 﨎 0;– le champ des charges superficielles つs; un calcul classique conduit à la relation つs = は/3 﨎 0;– le champ de cavité つcav (champ des dipôles dans la cavité); d’une façon générale, ce champ est nul dans les diélectriques de classe A. E cav est nul également dans les matériaux cristallins dont la maille a un centre de symétrie (halogénures alcalins par exemple). Bien que certaines propriétés physiques des cristaux cubiques ne soient pas isotropes (les propriétés de transport, de cohésion, d’absorption optique, etc., peuvent varier avec la direction cristalline), on peut montrer que le champ de cavité est nul dans ces cristaux, et par suite: Remarquons qu’il ne faut pas confondre la cavité fictive de Mossotti avec une cavité réelle. La figure 6 indique les différences entre les deux types de cavité.Relations de Clausius-Mossotti et de Lorentz-LorenzEn revenant à la définition de la polarisation:
Remarquons qu’il ne faut pas confondre la cavité fictive de Mossotti avec une cavité réelle. La figure 6 indique les différences entre les deux types de cavité.Relations de Clausius-Mossotti et de Lorentz-LorenzEn revenant à la définition de la polarisation: et en remplaçant le champ local つ par sa valeur, on trouve la polarisabilité relative à l’unité de volume:
et en remplaçant le champ local つ par sa valeur, on trouve la polarisabilité relative à l’unité de volume: Or, une molécule-gramme de masse M contient 類 molécules, 類 étant le nombre d’Avogadro ( 類 = 6,023... 1023). Si 福 désigne la densité du matériau, et M la masse moléculaire moyenne de ses constituants, on a donc: N = 福 類 /M , et par suite on peut écrire la polarisabilité «molaire» sous la forme:
Or, une molécule-gramme de masse M contient 類 molécules, 類 étant le nombre d’Avogadro ( 類 = 6,023... 1023). Si 福 désigne la densité du matériau, et M la masse moléculaire moyenne de ses constituants, on a donc: N = 福 類 /M , et par suite on peut écrire la polarisabilité «molaire» sous la forme: ce qui constitue l’importante relation de Clausius-Mossotti.La relation de Clausius-Mossotti, établie pour une polarisation statique, a été étendue au cas de la polarisation dynamique en particulier aux fréquences optiques, indépendamment par Hendrik A. Lorentz et Ludwig V. Lorenz. À ces fréquences, = n 2, n étant l’indice de réfraction optique, d’où:
ce qui constitue l’importante relation de Clausius-Mossotti.La relation de Clausius-Mossotti, établie pour une polarisation statique, a été étendue au cas de la polarisation dynamique en particulier aux fréquences optiques, indépendamment par Hendrik A. Lorentz et Ludwig V. Lorenz. À ces fréquences, = n 2, n étant l’indice de réfraction optique, d’où: Plus généralement, aux fréquences où la permittivité est une quantité complexe, on peut écrire:
Plus généralement, aux fréquences où la permittivité est une quantité complexe, on peut écrire: avec face="EU Domacr" 滑 = n 漣 jk .Application à la mesure des polarisabilités d’orientationLa polarisabilité totale 見 résulte de la polarisabilité électronique ou atomique 見 e et de la polarisabilité d’orientation 見 or:
avec face="EU Domacr" 滑 = n 漣 jk .Application à la mesure des polarisabilités d’orientationLa polarisabilité totale 見 résulte de la polarisabilité électronique ou atomique 見 e et de la polarisabilité d’orientation 見 or: Comme on le verra, ces deux composantes ont des temps de réponse différents. Aux hautes fréquences (fréquences de résonance), seule la composante 見 e intervient, et la permittivité correspondante est size=1秊. La formule de Clausius-Mossotti s’écrit alors:
Comme on le verra, ces deux composantes ont des temps de réponse différents. Aux hautes fréquences (fréquences de résonance), seule la composante 見 e intervient, et la permittivité correspondante est size=1秊. La formule de Clausius-Mossotti s’écrit alors: Aux fréquences inférieures aux fréquences de relaxation dipolaire, 見 or intervient également, et la permittivité correspondante est s 礪 size=1秊. La formule de Clausius-Mossotti s’écrit dans ce cas:
Aux fréquences inférieures aux fréquences de relaxation dipolaire, 見 or intervient également, et la permittivité correspondante est s 礪 size=1秊. La formule de Clausius-Mossotti s’écrit dans ce cas: M , 福 , s et size=1秊 étant connus, par la mesure, les relations (28) et (29) permettent de trouver 見 e et 見 or, et, par suite, le moment dipolaire des molécules, grâce à la formule de Langevin: 見 or = 猪 2/3 kT .Limite de validité de la formule de Clausius-MossottiLa formule du champ local n’est pas satisfaisante pour deux raisons.D’une part, elle est totalement fausse pour les matériaux polaires (c’est-à-dire à dipôles permanents), puisqu’elle prédit que tout matériau polaire, même isotrope, doit avoir une permittivité infinie si on abaisse suffisamment sa température. Cela tient à l’insuffisance du modèle de la cavité. En effet, en extrayant de la formule de Clausius-Mossotti, on obtient une fonction croissante de N 見 , qui tend vers l’infini si N 見 tend vers 3 﨎 0. Si le diélectrique est constitué de dipôles permanents, ces dipôles sont soumis aux sollicitations antagonistes du champ appliqué tendant à les orienter et de l’agitation thermique tendant à les désorienter. Ainsi, la polarisabilité apparente augmente quand la température diminue, et le terme N 見 peut devenir aussi grand que 3 﨎 0 si la température est assez basse, de sorte que le modèle prévoit, pour tout matériau polaire, une température critique à laquelle est infini, ce qui est, bien entendu, contraire à l’expérience.D’autre part, lorsqu’on considère les positions moyennes des dipôles, comme on le fait pour calculer E cav, on suppose que les mouvements thermiques des différents dipôles sont totalement indépendants. Il n’en est rien, du fait des interactions dipôle-dipôle. L’orientation moyenne de chaque dipôle, considéré séparément, est effectivement parallèle à celle du champ extérieur, mais l’interaction qui existe à chaque instant entre les dipôles voisins empêche le champ de cavité d’être nul, de sorte que la formule du champ local n’est qu’une approximation, même lorsqu’il s’agit de dipôles induits.Extensions théoriquesLa relation de Clausius-Mossotti est une excellente approximation pour les corps non polaires (même condensés) et même pour les gaz polaires; mais, pour les raisons qu’on vient d’exposer, elle ne peut pas s’appliquer aux corps polaires condensés.Le problème des corps polaires condensés a fait l’objet de très nombreux travaux théoriques. Le premier modèle qui ait évité l’écueil du modèle de Clausius-Mossotti a été fourni par Lars Onsager ; il est développé dans la plupart des références bibliographiques.La différence essentielle entre la théorie de Clausius-Mossotti et celle d’Onsager réside dans la nature de la cavité considérée.Dans la théorie de Clausius-Mossotti, la cavité est grande par rapport aux dimensions moléculaires, et la polarisation de la cavité est calculée dans l’hypothèse où les lignes de force restent parallèles au champ appliqué.Onsager considère une cavité réelle, constituée par une vacuole de dimension moléculaire, qui dévie les lignes de force à l’extérieur. Il calcule, à partir de l’équation de Laplace, le champ à l’intérieur d’une telle cavité, puis il tient compte de la polarisation à l’intérieur en calculant au centre de la cavité le champ de «réaction» qui provient de la polarisation de la cavité par un dipôle fictif situé en son centre.La théorie d’Onsager est elle-même une approximation: elle ne tient pas compte de l’interaction directionnelle spécifique entre molécules, étudiée en particulier par Fröhlich. Si 﨏 ij désigne l’angle entre les orientations de deux molécules i et j , la moyenne de 﨏 ij sur l’ensemble des couples est le plus souvent nulle. Toutefois, il existe parfois une interaction directionnelle entre les molécules. C’est le cas, par exemple, de l’acide cyanhydrique, dont les molécules sont associées par des liaisons hydrogènes pour former des chaînes rectilignes finies sans qu’il s’agisse pourtant d’un polymère stable, mais d’un polymère dit «labile».Dans ce cas, la valeur moyenne de cos 﨏 ij n’est pas nulle, et l’on peut définir pour la molécule i un paramètre de corrélation:
M , 福 , s et size=1秊 étant connus, par la mesure, les relations (28) et (29) permettent de trouver 見 e et 見 or, et, par suite, le moment dipolaire des molécules, grâce à la formule de Langevin: 見 or = 猪 2/3 kT .Limite de validité de la formule de Clausius-MossottiLa formule du champ local n’est pas satisfaisante pour deux raisons.D’une part, elle est totalement fausse pour les matériaux polaires (c’est-à-dire à dipôles permanents), puisqu’elle prédit que tout matériau polaire, même isotrope, doit avoir une permittivité infinie si on abaisse suffisamment sa température. Cela tient à l’insuffisance du modèle de la cavité. En effet, en extrayant de la formule de Clausius-Mossotti, on obtient une fonction croissante de N 見 , qui tend vers l’infini si N 見 tend vers 3 﨎 0. Si le diélectrique est constitué de dipôles permanents, ces dipôles sont soumis aux sollicitations antagonistes du champ appliqué tendant à les orienter et de l’agitation thermique tendant à les désorienter. Ainsi, la polarisabilité apparente augmente quand la température diminue, et le terme N 見 peut devenir aussi grand que 3 﨎 0 si la température est assez basse, de sorte que le modèle prévoit, pour tout matériau polaire, une température critique à laquelle est infini, ce qui est, bien entendu, contraire à l’expérience.D’autre part, lorsqu’on considère les positions moyennes des dipôles, comme on le fait pour calculer E cav, on suppose que les mouvements thermiques des différents dipôles sont totalement indépendants. Il n’en est rien, du fait des interactions dipôle-dipôle. L’orientation moyenne de chaque dipôle, considéré séparément, est effectivement parallèle à celle du champ extérieur, mais l’interaction qui existe à chaque instant entre les dipôles voisins empêche le champ de cavité d’être nul, de sorte que la formule du champ local n’est qu’une approximation, même lorsqu’il s’agit de dipôles induits.Extensions théoriquesLa relation de Clausius-Mossotti est une excellente approximation pour les corps non polaires (même condensés) et même pour les gaz polaires; mais, pour les raisons qu’on vient d’exposer, elle ne peut pas s’appliquer aux corps polaires condensés.Le problème des corps polaires condensés a fait l’objet de très nombreux travaux théoriques. Le premier modèle qui ait évité l’écueil du modèle de Clausius-Mossotti a été fourni par Lars Onsager ; il est développé dans la plupart des références bibliographiques.La différence essentielle entre la théorie de Clausius-Mossotti et celle d’Onsager réside dans la nature de la cavité considérée.Dans la théorie de Clausius-Mossotti, la cavité est grande par rapport aux dimensions moléculaires, et la polarisation de la cavité est calculée dans l’hypothèse où les lignes de force restent parallèles au champ appliqué.Onsager considère une cavité réelle, constituée par une vacuole de dimension moléculaire, qui dévie les lignes de force à l’extérieur. Il calcule, à partir de l’équation de Laplace, le champ à l’intérieur d’une telle cavité, puis il tient compte de la polarisation à l’intérieur en calculant au centre de la cavité le champ de «réaction» qui provient de la polarisation de la cavité par un dipôle fictif situé en son centre.La théorie d’Onsager est elle-même une approximation: elle ne tient pas compte de l’interaction directionnelle spécifique entre molécules, étudiée en particulier par Fröhlich. Si 﨏 ij désigne l’angle entre les orientations de deux molécules i et j , la moyenne de 﨏 ij sur l’ensemble des couples est le plus souvent nulle. Toutefois, il existe parfois une interaction directionnelle entre les molécules. C’est le cas, par exemple, de l’acide cyanhydrique, dont les molécules sont associées par des liaisons hydrogènes pour former des chaînes rectilignes finies sans qu’il s’agisse pourtant d’un polymère stable, mais d’un polymère dit «labile».Dans ce cas, la valeur moyenne de cos 﨏 ij n’est pas nulle, et l’on peut définir pour la molécule i un paramètre de corrélation: et la formule d’Onsager doit être affectée d’un coefficient de corrélation moyen g 礪 1.D’autres chercheurs, tels Kirkwood, Fuoss, etc., ont proposé des améliorations aux formules précédentes, le plus souvent, hélas! aux dépens de la simplicité.Le tableau 2 résume les relations les plus simples entre la polarisabilité molaire d’orientation et les permittivités mesurées. Toutes ces formules sont de la forme:
et la formule d’Onsager doit être affectée d’un coefficient de corrélation moyen g 礪 1.D’autres chercheurs, tels Kirkwood, Fuoss, etc., ont proposé des améliorations aux formules précédentes, le plus souvent, hélas! aux dépens de la simplicité.Le tableau 2 résume les relations les plus simples entre la polarisabilité molaire d’orientation et les permittivités mesurées. Toutes ces formules sont de la forme: la quantité A est donnée d’après les différentes théories (tabl. 2).7. ÉlectrostrictionL’électrostriction est un phénomène relatif à la déformation d’un milieu diélectrique non chargé causée par la présence d’un champ électrique.Fluides isotropesDans les fluides isotropes, l’électrostriction tend à déplacer le matériau pour l’accumuler dans des zones de champ fort, y provoquant une surpression. Pour calculer cette surpression, il faut appliquer la méthode d’Helmholtz, qui consiste à écrire que le travail des forces de déformation est compensé par la variation d’énergie électrostatique.Dans le cas particulier d’un fluide quasi incompressible de densité 福 et de permittivité 﨎 , la méthode d’Helmholtz conduit à une expression simple de la surpression 蓮p entre une zone où le champ est E et une zone équivalente, du point de vue hydrostatique, où le champ est nul:
la quantité A est donnée d’après les différentes théories (tabl. 2).7. ÉlectrostrictionL’électrostriction est un phénomène relatif à la déformation d’un milieu diélectrique non chargé causée par la présence d’un champ électrique.Fluides isotropesDans les fluides isotropes, l’électrostriction tend à déplacer le matériau pour l’accumuler dans des zones de champ fort, y provoquant une surpression. Pour calculer cette surpression, il faut appliquer la méthode d’Helmholtz, qui consiste à écrire que le travail des forces de déformation est compensé par la variation d’énergie électrostatique.Dans le cas particulier d’un fluide quasi incompressible de densité 福 et de permittivité 﨎 , la méthode d’Helmholtz conduit à une expression simple de la surpression 蓮p entre une zone où le champ est E et une zone équivalente, du point de vue hydrostatique, où le champ est nul: Pour un fluide non polaire , on peut relier la permittivité à la densité 福 par la formule de Clausius-Mossotti établie plus haut:
Pour un fluide non polaire , on peut relier la permittivité à la densité 福 par la formule de Clausius-Mossotti établie plus haut: où C est une constante:
où C est une constante: Par différenciation, cette relation devient:
Par différenciation, cette relation devient: et, en combinant (30) et (31), on obtient:
et, en combinant (30) et (31), on obtient: Supposons, par exemple, = 2 (liquide non polaire). Dans ce cas, la pression (en newtons par mètre carré) est liée au champ (en volts par mètre) par la relation:
Supposons, par exemple, = 2 (liquide non polaire). Dans ce cas, la pression (en newtons par mètre carré) est liée au champ (en volts par mètre) par la relation: soit, en unités usuelles:
soit, en unités usuelles: On voit que le champ doit être de l’ordre de 108 Vm-1 pour que la surpression d’électrostriction soit de l’ordre du bar. L’effet, variant comme le carré du champ E , est très petit en champ faible.Cas des cristauxTous les cristaux présentent une électrostriction quadratique comme les fluides, à ceci près que le «cœfficient d’électrostriction», qui relie le carré de la polarisation à la déformation locale, n’est plus un scalaire, mais un tenseur du quatrième ordre.Certains cristaux à symétrie axiale sont piézo-électriques (cf. PIÉZO-ÉLECTRICITÉ). Un champ électrique appliqué à un tel cristal le déforme, en raison de la réversibilité de l’effet piézo-électrique. Toutefois, il ne faut pas confondre cet effet linéaire avec l’électrostriction, qui est toujours quadratique.8. Variation de la permittivité avec la fréquenceDivers types de charges et groupements de chargesDans le but d’étudier comment varie la permittivité d’un diélectrique avec la fréquence de mesure, il faut d’abord faire le bilan des divers types de charges et groupements de charges soumis à l’action du champ. On distingue:– les électrons des couches internes , fortement liés aux noyaux atomiques;– les électrons des couches externes , ou électrons de valence dans les atomes ou les molécules;– les électrons libres ou électrons de conduction (en petit nombre dans les diélectriques);– les ions liés à d’autres ions de charge opposée, dans les molécules ou les cristaux (exemple: Cl- H+); ces paires d’ions forment des dipôles permanents ;– les ions libres , tels que ceux des électrolytes et des cristaux ioniques (exemple: ions K+ excédentaires dans KCl). Ces ions peuvent être associés à des dipôles permanents (exemple: OH-);– les dipôles induits par le champ appliqué; par exemple, une molécule de benzène, de moment dipolaire nul en l’absence de champ, se polarise et acquiert un dipôle induit dans un champ électrique, à cause d’une redistribution des électrons de valence (notons qu’un champ électrique modifie le moment dipolaire d’un dipôle permanent);– les multipôles ; par exemple, deux dipôles identiques associés et antiparallèles ont un moment dipolaire nul: ils forment un quadrupôle.Tous ces types de charges ou de groupements de charges sont perturbés par le champ électrique. Les charges libres se déplacent dans la direction du champ si elles sont positives, ou vice versa.Les charges liées tendent à se déplacer (et en fait se déplacent légèrement), mais sont maintenues par les forces de rappel, qui augmentent très vite avec le déplacement.Les dipôles tendent à s’orienter dans la position d’énergie potentielle minimale, c’est-à-dire dans la direction du champ, et, en fait, tous les dipôles s’aligneraient si l’agitation thermique ne s’opposait à cette orientation (la théorie de Langevin pour le paramagnétisme est formellement identique).Enfin, les multipôles ne s’orientent pas, mais se déforment légèrement.Si on annule brusquement une contrainte électrique appliquée, toutes les charges et tous les groupements de charges évoluent vers l’état initial. Les dipôles induits se dépolarisent et s’annulent, l’anisotropie due à l’orientation des dipôles permanents disparaît, les charges libres accumulées aux électrodes diffusent de manière à annuler les gradients de concentration, etc.Chaque type de charge ou de groupement se déplace, ou se déforme, avec une vitesse qui lui est propre, et qui dépend d’une part de la masse des particules en jeu et d’autre part des forces de rappel ou de frottement (fig. 7).Ainsi, les électrons des couches internes des atomes, liés au noyau par des forces énormes, ont des mouvements propres extrêmement rapides, dont la période, de l’ordre de 10-19 seconde, correspond à celle du rayonnement X. Par suite, un rayonnement électromagnétique d’une fréquence supérieure (ou d’une période inférieure), trop «rapide» pour amorcer les vibrations électroniques, n’a aucun effet polarisant, et la permittivité relative des matériaux à ces fréquences est égale, ou très légèrement inférieure à celle du vide (point 1 de la fig. 7).Si on abaisse la fréquence du rayonnement incident au-dessous de la fréquence de résonance des électrons internes, ces électrons ont, en quelque sorte, le temps d’obéir à la sollicitation du vecteur électrique du champ; ils oscillent avec le champ appliqué et polarisent le matériau à la fréquence correspondante, la permittivité relative devient alors légèrement supérieure à l’unité (point 2 de la fig. 7).Si on abaisse maintenant la fréquence au-dessous de la fréquence propre des électrons de valence, qui se situe dans le visible ou l’ultraviolet ( = 0,1 à 1 猪m, f 黎 3.1014 à 3.1015 Hz), ces électrons se mettent à vibrer avec le champ appliqué, et une nouvelle polarisation contribue à la permittivité, qui augmente à nouveau (point 3 de la fig. 7).Le même phénomène se produit aux fréquences correspondant aux vibrations atomiques dans les molécules ou les cristaux, qui se situent dans l’infrarouge ( = 10 à 100 猪m, soit f = 3.1012 à 3.1013 Hz: point 4 de la fig. 7).Tous les phénomènes décrits jusqu’ici sont des déplacements de charges rappelées vers leur position d’équilibre par des forces de rappel proportionnelles à l’amplitude du déplacement. Les accélérations en jeu sont donc très importantes, et, même si les masses sont faibles, les termes relatifs à l’inertie dans les équations cinétiques sont importants.Il s’agit de résonances , plus ou moins amorties (et élargies) sous l’effet des forces de frottement relativement faibles dues essentiellement à la dissipation d’énergie par les charges accélérées (rayonnement de freinage). Ces phénomènes de résonance sont d’intérêt majeur pour les diélectriciens (cf. MÉCANIQUE ANALYTIQUE, SPECTROSCOPIE, OPTIQUE).Si on continue à abaisser la fréquence du champ appliqué au-dessous de celle des vibrations atomiques, les phénomènes prennent une forme nouvelle. Considérons, par exemple, des dipôles orientés dans un champ appliqué, et supposons que ce champ s’annule brusquement. En l’absence de forces de rappel élastiques, les seules forces tendant à ramener les dipôles dans une orientation quelconque, c’est-à-dire à redonner au système l’isotropie qu’il avait avant l’application du champ, sont les collisions avec les particules voisines, qui transmettent aux dipôles une partie de leur énergie cinétique d’origine thermique.Contrairement à ce qui caractérise les résonances, les termes de frottement sont ici prédominants sur les termes relatifs à l’inertie. Il s’agit de phénomènes de relaxation .Les phénomènes de relaxation dipolaire se produisent en général entre 107 et 1010 hertz, mais on peut les mettre en évidence à des fréquences extérieures à cet intervalle. On conçoit en effet, d’une part, que ces relaxations sont beaucoup plus étalées dans le spectre que les résonances étroitement localisées autour des fréquences propres, et, d’autre part, que la fréquence centrale de la relaxation dépend très fortement des propriétés physiques du milieu, en particulier de sa viscosité.Comme précédemment, les relaxations contribuèrent à la permittivité du milieu, si la fréquence de mesure est inférieure à la fréquence centrale de relaxation (point 5 sur la fig. 7).Aux basses et très basses fréquences, pouvant descendre jusqu’à 10-4 hertz, apparaissent des phénomènes de relaxation mettant en jeu l’ensemble de l’échantillon. Ce sont essentiellement les phénomènes de relaxation de la concentration des charges libres ou charges d’espace, dont l’effet Maxwell-Wagner est un cas particulier et qui sont encore assez mal connus en raison des difficultés expérimentales et de l’influence grandissante des électrodes aux très basses fréquences.Ces effets peuvent accroître considérablement la permittivité à très basse fréquence de certains matériaux peu conducteurs, où la mobilité des porteurs est très faible (point 6 sur la fig. 7).9. Relaxation dipolaireOn appelle relaxation un retour vers l’équilibre thermodynamique d’un ensemble de particules en interaction (par exemple, dipôles), après suppression d’une perturbation (par exemple, champ électrique).Modèle de DebyeOn applique, à l’instant t = 0, sur le matériau électrique un champ électrique つ, dont le temps d’établissement est supposé court par rapport aux phénomènes de relaxation envisagés, mais pas assez court pour que les très faibles retards causés par l’inertie des particules résonantes (électrons ou atomes) doivent être pris en considération ( 礪 10-10 s.). En d’autres termes, on suppose qu’à tout instant la polarisation est la somme d’une composante P res = P 4, correspondant aux phénomènes de résonance qui apparaissent en moins de 10-10 seconde après l’application du champ, et d’une composante P retardée, correspondant aux phénomènes de relaxation dipolaire jusqu’à 10-4 seconde:
On voit que le champ doit être de l’ordre de 108 Vm-1 pour que la surpression d’électrostriction soit de l’ordre du bar. L’effet, variant comme le carré du champ E , est très petit en champ faible.Cas des cristauxTous les cristaux présentent une électrostriction quadratique comme les fluides, à ceci près que le «cœfficient d’électrostriction», qui relie le carré de la polarisation à la déformation locale, n’est plus un scalaire, mais un tenseur du quatrième ordre.Certains cristaux à symétrie axiale sont piézo-électriques (cf. PIÉZO-ÉLECTRICITÉ). Un champ électrique appliqué à un tel cristal le déforme, en raison de la réversibilité de l’effet piézo-électrique. Toutefois, il ne faut pas confondre cet effet linéaire avec l’électrostriction, qui est toujours quadratique.8. Variation de la permittivité avec la fréquenceDivers types de charges et groupements de chargesDans le but d’étudier comment varie la permittivité d’un diélectrique avec la fréquence de mesure, il faut d’abord faire le bilan des divers types de charges et groupements de charges soumis à l’action du champ. On distingue:– les électrons des couches internes , fortement liés aux noyaux atomiques;– les électrons des couches externes , ou électrons de valence dans les atomes ou les molécules;– les électrons libres ou électrons de conduction (en petit nombre dans les diélectriques);– les ions liés à d’autres ions de charge opposée, dans les molécules ou les cristaux (exemple: Cl- H+); ces paires d’ions forment des dipôles permanents ;– les ions libres , tels que ceux des électrolytes et des cristaux ioniques (exemple: ions K+ excédentaires dans KCl). Ces ions peuvent être associés à des dipôles permanents (exemple: OH-);– les dipôles induits par le champ appliqué; par exemple, une molécule de benzène, de moment dipolaire nul en l’absence de champ, se polarise et acquiert un dipôle induit dans un champ électrique, à cause d’une redistribution des électrons de valence (notons qu’un champ électrique modifie le moment dipolaire d’un dipôle permanent);– les multipôles ; par exemple, deux dipôles identiques associés et antiparallèles ont un moment dipolaire nul: ils forment un quadrupôle.Tous ces types de charges ou de groupements de charges sont perturbés par le champ électrique. Les charges libres se déplacent dans la direction du champ si elles sont positives, ou vice versa.Les charges liées tendent à se déplacer (et en fait se déplacent légèrement), mais sont maintenues par les forces de rappel, qui augmentent très vite avec le déplacement.Les dipôles tendent à s’orienter dans la position d’énergie potentielle minimale, c’est-à-dire dans la direction du champ, et, en fait, tous les dipôles s’aligneraient si l’agitation thermique ne s’opposait à cette orientation (la théorie de Langevin pour le paramagnétisme est formellement identique).Enfin, les multipôles ne s’orientent pas, mais se déforment légèrement.Si on annule brusquement une contrainte électrique appliquée, toutes les charges et tous les groupements de charges évoluent vers l’état initial. Les dipôles induits se dépolarisent et s’annulent, l’anisotropie due à l’orientation des dipôles permanents disparaît, les charges libres accumulées aux électrodes diffusent de manière à annuler les gradients de concentration, etc.Chaque type de charge ou de groupement se déplace, ou se déforme, avec une vitesse qui lui est propre, et qui dépend d’une part de la masse des particules en jeu et d’autre part des forces de rappel ou de frottement (fig. 7).Ainsi, les électrons des couches internes des atomes, liés au noyau par des forces énormes, ont des mouvements propres extrêmement rapides, dont la période, de l’ordre de 10-19 seconde, correspond à celle du rayonnement X. Par suite, un rayonnement électromagnétique d’une fréquence supérieure (ou d’une période inférieure), trop «rapide» pour amorcer les vibrations électroniques, n’a aucun effet polarisant, et la permittivité relative des matériaux à ces fréquences est égale, ou très légèrement inférieure à celle du vide (point 1 de la fig. 7).Si on abaisse la fréquence du rayonnement incident au-dessous de la fréquence de résonance des électrons internes, ces électrons ont, en quelque sorte, le temps d’obéir à la sollicitation du vecteur électrique du champ; ils oscillent avec le champ appliqué et polarisent le matériau à la fréquence correspondante, la permittivité relative devient alors légèrement supérieure à l’unité (point 2 de la fig. 7).Si on abaisse maintenant la fréquence au-dessous de la fréquence propre des électrons de valence, qui se situe dans le visible ou l’ultraviolet ( = 0,1 à 1 猪m, f 黎 3.1014 à 3.1015 Hz), ces électrons se mettent à vibrer avec le champ appliqué, et une nouvelle polarisation contribue à la permittivité, qui augmente à nouveau (point 3 de la fig. 7).Le même phénomène se produit aux fréquences correspondant aux vibrations atomiques dans les molécules ou les cristaux, qui se situent dans l’infrarouge ( = 10 à 100 猪m, soit f = 3.1012 à 3.1013 Hz: point 4 de la fig. 7).Tous les phénomènes décrits jusqu’ici sont des déplacements de charges rappelées vers leur position d’équilibre par des forces de rappel proportionnelles à l’amplitude du déplacement. Les accélérations en jeu sont donc très importantes, et, même si les masses sont faibles, les termes relatifs à l’inertie dans les équations cinétiques sont importants.Il s’agit de résonances , plus ou moins amorties (et élargies) sous l’effet des forces de frottement relativement faibles dues essentiellement à la dissipation d’énergie par les charges accélérées (rayonnement de freinage). Ces phénomènes de résonance sont d’intérêt majeur pour les diélectriciens (cf. MÉCANIQUE ANALYTIQUE, SPECTROSCOPIE, OPTIQUE).Si on continue à abaisser la fréquence du champ appliqué au-dessous de celle des vibrations atomiques, les phénomènes prennent une forme nouvelle. Considérons, par exemple, des dipôles orientés dans un champ appliqué, et supposons que ce champ s’annule brusquement. En l’absence de forces de rappel élastiques, les seules forces tendant à ramener les dipôles dans une orientation quelconque, c’est-à-dire à redonner au système l’isotropie qu’il avait avant l’application du champ, sont les collisions avec les particules voisines, qui transmettent aux dipôles une partie de leur énergie cinétique d’origine thermique.Contrairement à ce qui caractérise les résonances, les termes de frottement sont ici prédominants sur les termes relatifs à l’inertie. Il s’agit de phénomènes de relaxation .Les phénomènes de relaxation dipolaire se produisent en général entre 107 et 1010 hertz, mais on peut les mettre en évidence à des fréquences extérieures à cet intervalle. On conçoit en effet, d’une part, que ces relaxations sont beaucoup plus étalées dans le spectre que les résonances étroitement localisées autour des fréquences propres, et, d’autre part, que la fréquence centrale de la relaxation dépend très fortement des propriétés physiques du milieu, en particulier de sa viscosité.Comme précédemment, les relaxations contribuèrent à la permittivité du milieu, si la fréquence de mesure est inférieure à la fréquence centrale de relaxation (point 5 sur la fig. 7).Aux basses et très basses fréquences, pouvant descendre jusqu’à 10-4 hertz, apparaissent des phénomènes de relaxation mettant en jeu l’ensemble de l’échantillon. Ce sont essentiellement les phénomènes de relaxation de la concentration des charges libres ou charges d’espace, dont l’effet Maxwell-Wagner est un cas particulier et qui sont encore assez mal connus en raison des difficultés expérimentales et de l’influence grandissante des électrodes aux très basses fréquences.Ces effets peuvent accroître considérablement la permittivité à très basse fréquence de certains matériaux peu conducteurs, où la mobilité des porteurs est très faible (point 6 sur la fig. 7).9. Relaxation dipolaireOn appelle relaxation un retour vers l’équilibre thermodynamique d’un ensemble de particules en interaction (par exemple, dipôles), après suppression d’une perturbation (par exemple, champ électrique).Modèle de DebyeOn applique, à l’instant t = 0, sur le matériau électrique un champ électrique つ, dont le temps d’établissement est supposé court par rapport aux phénomènes de relaxation envisagés, mais pas assez court pour que les très faibles retards causés par l’inertie des particules résonantes (électrons ou atomes) doivent être pris en considération ( 礪 10-10 s.). En d’autres termes, on suppose qu’à tout instant la polarisation est la somme d’une composante P res = P 4, correspondant aux phénomènes de résonance qui apparaissent en moins de 10-10 seconde après l’application du champ, et d’une composante P retardée, correspondant aux phénomènes de relaxation dipolaire jusqu’à 10-4 seconde: Immédiatement après l’application du champ, on a P (0+) = P 4. Puis P augmente régulièrement jusqu’à une valeur asymptotique P (face=F0019 秊) = P 5 (les indices 4 et 5 sont relatifs aux points 4 et 5 de la fig. 7).Supposons que le taux d’augmentation de P soit proportionnel à la différence entre sa valeur limite P (face=F0019 秊) = P 5 漣 P 4 et la valeur instantanée P (t ):
Immédiatement après l’application du champ, on a P (0+) = P 4. Puis P augmente régulièrement jusqu’à une valeur asymptotique P (face=F0019 秊) = P 5 (les indices 4 et 5 sont relatifs aux points 4 et 5 de la fig. 7).Supposons que le taux d’augmentation de P soit proportionnel à la différence entre sa valeur limite P (face=F0019 秊) = P 5 漣 P 4 et la valeur instantanée P (t ): où 精 désigne un temps caractéristique du matériau qu’on appelle «temps de relaxation». Par intégration, on en déduit P (t ) et:
où 精 désigne un temps caractéristique du matériau qu’on appelle «temps de relaxation». Par intégration, on en déduit P (t ) et: On voit que la polarisation résultant d’un échelon de tension appliquée varie exponentiellement de P 4 à P 5, avec la constante de temps 精 . Pour la relaxation considérée l’indice 5 correspondant aux «basses fréquences» est souvent remplacé par l’indice s (statique), et l’indice 4 par l’indice 秊 (pour fréquence infinie).Soumettons maintenant le matériau à un champ alternatif de pulsation 諸 . P 4 change de signe au début de chaque alternance. Nous nous intéresserons à la solution permanente de l’équation (33) en régime sinusoïdal, et nous admettons donc que P varie sinusoïdalement à la pulsation 諸 du champ appliqué.
On voit que la polarisation résultant d’un échelon de tension appliquée varie exponentiellement de P 4 à P 5, avec la constante de temps 精 . Pour la relaxation considérée l’indice 5 correspondant aux «basses fréquences» est souvent remplacé par l’indice s (statique), et l’indice 4 par l’indice 秊 (pour fréquence infinie).Soumettons maintenant le matériau à un champ alternatif de pulsation 諸 . P 4 change de signe au début de chaque alternance. Nous nous intéresserons à la solution permanente de l’équation (33) en régime sinusoïdal, et nous admettons donc que P varie sinusoïdalement à la pulsation 諸 du champ appliqué. d’où: P( 諸 ) = P 4 + (P 5 漣 P 4)/(1 + j 諸 精).Notons que le trop bref passage du régime en échelon au régime harmonique peut se justifier rigoureusement à l’aide de la théorie de la réponse indicielle.En utilisant la relation entre P et E on peut mettre la permittivité complexe sous la forme:
d’où: P( 諸 ) = P 4 + (P 5 漣 P 4)/(1 + j 諸 精).Notons que le trop bref passage du régime en échelon au régime harmonique peut se justifier rigoureusement à l’aide de la théorie de la réponse indicielle.En utilisant la relation entre P et E on peut mettre la permittivité complexe sous la forme: où 精 = [( 﨎 s + 2 﨎 0)/( 﨎 秊 + 2 﨎 0)] 精 . De la relation (34), appelée relation de Debye, on déduit 﨎 ( 諸 ) et 﨎 ( 諸 ) par séparation des parties réelle et imaginaire.En régime sinusoïdal, la relaxation dipolaire se manifeste par un retard entre le champ appliqué et la polarisation. Ce retard est lié aux perturbations exercées par les molécules en agitation thermique, qui freinent l’orientation (ou la désorientation) des dipôles, ou l’accumulation de charges, dans un champ appliqué. Ces phénomènes sont irréversibles et correspondent à une dissipation d’énergie.Représentations graphiquesOn peut représenter séparément les composantes 﨎 et 﨎 de 﨎 en fonction de 諸 . On obtient pour 﨎 ( 諸 ) une courbe analogue à celle qui joint les points 4 et 5 de la figure 7. Quant à 﨎 ( 諸 ), il est proportionnel à la dérivée de 﨎 ( 諸 ) (courbe en cloche).Afin de préciser la nature d’un phénomène dispersif, il est particulièrement intéressant d’utiliser le diagramme 﨎 ( 﨎 ) (diagramme d’Argand). Un simple recours à l’inversion géométrique montre que le diagramme d’Argand de la relation de Debye est un demi-cercle centré en ( 﨎 s + 﨎 size=1秊)/2 sur l’axe 﨎 . C’est le demi-cercle de Cole (fig. 8).Par suite, si les points expérimentaux sont répartis sur un demi-cercle de Cole, le mécanisme de dispersion est une relaxation dipolaire de type Debye, et le temps de relaxation est donné par l’inverse de la pulsation correspondant au sommet du demi-cercle.Distribution de temps de relaxationUne relaxation dipolaire de Debye est obtenue avec de nombreux liquides diélectriques, en particulier les alcools (Davidson et Cole, 1951), mais la plupart des spectres qui leur correspondent ne sont pas exactement conformes à la théorie de Debye; ils sont en général intérieurs au demi-cercle de Cole. Cette différence a été interprétée comme une conséquence de l’existence, dans un diélectrique condensé, de plusieurs types de dipôles et, pour chaque type, de plusieurs mouvements possibles et de plusieurs configurations: il existe alors, non pas un seul, mais tout un ensemble de temps de relaxation. On est même conduit à admettre que chaque entité dipolaire a, à un instant donné, un temps de relaxation différent de celui des entités voisines, de sorte que si l’on considère un grand nombre de dipôles, on est conduit à la notion de distribution de temps de relaxation. Si la proportion des dipôles dont le temps de relaxation compris entre 精 et 精 + d 精 est f ( 精 ) d 精 , f ( 精 ) est la fonction de distribution (normalisée) du temps de relaxation.À une fonction de distribution donnée ( 精), on peut associer une permittivité:
où 精 = [( 﨎 s + 2 﨎 0)/( 﨎 秊 + 2 﨎 0)] 精 . De la relation (34), appelée relation de Debye, on déduit 﨎 ( 諸 ) et 﨎 ( 諸 ) par séparation des parties réelle et imaginaire.En régime sinusoïdal, la relaxation dipolaire se manifeste par un retard entre le champ appliqué et la polarisation. Ce retard est lié aux perturbations exercées par les molécules en agitation thermique, qui freinent l’orientation (ou la désorientation) des dipôles, ou l’accumulation de charges, dans un champ appliqué. Ces phénomènes sont irréversibles et correspondent à une dissipation d’énergie.Représentations graphiquesOn peut représenter séparément les composantes 﨎 et 﨎 de 﨎 en fonction de 諸 . On obtient pour 﨎 ( 諸 ) une courbe analogue à celle qui joint les points 4 et 5 de la figure 7. Quant à 﨎 ( 諸 ), il est proportionnel à la dérivée de 﨎 ( 諸 ) (courbe en cloche).Afin de préciser la nature d’un phénomène dispersif, il est particulièrement intéressant d’utiliser le diagramme 﨎 ( 﨎 ) (diagramme d’Argand). Un simple recours à l’inversion géométrique montre que le diagramme d’Argand de la relation de Debye est un demi-cercle centré en ( 﨎 s + 﨎 size=1秊)/2 sur l’axe 﨎 . C’est le demi-cercle de Cole (fig. 8).Par suite, si les points expérimentaux sont répartis sur un demi-cercle de Cole, le mécanisme de dispersion est une relaxation dipolaire de type Debye, et le temps de relaxation est donné par l’inverse de la pulsation correspondant au sommet du demi-cercle.Distribution de temps de relaxationUne relaxation dipolaire de Debye est obtenue avec de nombreux liquides diélectriques, en particulier les alcools (Davidson et Cole, 1951), mais la plupart des spectres qui leur correspondent ne sont pas exactement conformes à la théorie de Debye; ils sont en général intérieurs au demi-cercle de Cole. Cette différence a été interprétée comme une conséquence de l’existence, dans un diélectrique condensé, de plusieurs types de dipôles et, pour chaque type, de plusieurs mouvements possibles et de plusieurs configurations: il existe alors, non pas un seul, mais tout un ensemble de temps de relaxation. On est même conduit à admettre que chaque entité dipolaire a, à un instant donné, un temps de relaxation différent de celui des entités voisines, de sorte que si l’on considère un grand nombre de dipôles, on est conduit à la notion de distribution de temps de relaxation. Si la proportion des dipôles dont le temps de relaxation compris entre 精 et 精 + d 精 est f ( 精 ) d 精 , f ( 精 ) est la fonction de distribution (normalisée) du temps de relaxation.À une fonction de distribution donnée ( 精), on peut associer une permittivité: réciproquement, à un spectre d’Argand donné, on peut en principe, moyennant certaines réserves, faire correspondre une fonction de distribution.Se fondant sur l’observation que le diagramme d’Argand ( 﨎 , 﨎 ) est souvent un arc de cercle aplati, donc centré au-dessous de l’axe 﨎 , K. S. Cole et R. H. Cole ont proposé, pour interpréter ce spectre, une formule de Debye modifiée:
réciproquement, à un spectre d’Argand donné, on peut en principe, moyennant certaines réserves, faire correspondre une fonction de distribution.Se fondant sur l’observation que le diagramme d’Argand ( 﨎 , 﨎 ) est souvent un arc de cercle aplati, donc centré au-dessous de l’axe 﨎 , K. S. Cole et R. H. Cole ont proposé, pour interpréter ce spectre, une formule de Debye modifiée: où h est un paramètre qui caractérise l’aplatissement du diagramme.Pour h = 0, on retrouve la relation de Debye.Pour 0 諒 h 諒 1, le diagramme est bien un arc de cercle centré au-dessous de l’axe 﨎 , et la distribution f ( 精 ) correspondante a été calculée.Davidson et Cole ont effectué un travail analogue pour un diagramme en forme de demi-poire, incliné sur l’axe à 﨎 size=1秊, mais perpendiculaire à 﨎 s, tel qu’on l’observe souvent expérimentalement. Ces travaux sont fondamentaux, mais leur aspect macroscopique les rend peu exploitables.Depuis les développements de la mécanique statistique, et en particulier des travaux de R. Kubo, l’étude de la relaxation diélectrique en phase liquide a été entreprise à une échelle microscopique, et les travaux de S. H. Glarum ont ouvert une voie nouvelle vers l’interprétation des structures dipolaires en interaction. D’autre part, l’existence d’absorptions anormales a pu être attribuée aux effets d’inertie, qui peuvent devenir importants en haute fréquence. Notons enfin que des chercheurs de plusieurs universités françaises contribuent brillamment au développement de ces théories.10. Relaxation de charge d’espaceUn autre type important de relaxation est associé à l’accumulation de charges au voisinage d’électrodes plus ou moins bloquantes (c’est-à-dire plus ou moins impropres à décharger les ions arrivant à leur contact) sous l’influence combinée du champ, qui tend à accumuler les charges aux électrodes, et de l’agitation thermique qui, sous la forme du facteur de diffusion, s’oppose à cette accumulation.Considérons, par exemple, une plaque diélectrique d’un matériau de permittivité 﨎 contenant des charges libres entre électrodes bloquantes.Supposons que les charges des deux signes ont la même mobilité 猪 . Lorsqu’un champ permanent est appliqué entre les électrodes, les charges d’un même signe migrent vers l’électrode de signe opposé; cette migration est limitée par la diffusion, qui s’oppose à l’accumulation des charges. À l’état d’équilibre le matériau est macroscopiquement polarisé, et il contient une densité de charge 福 (x ), qui dépend de la distance x du point considéré aux électrodes. En d’autres termes, le matériau est équivalent à un dipôle (fig. 9). Si on inverse le sens du champ appliqué, la courbe de répartition des densités de charge évolue lentement vers la courbe b , symétrique de la courbe a par rapport à Ox, et, après un délai lié à la diffusion des charges, la configuration finale du matériau est équivalente à un dipôle opposé (fig. 9 b). Ce renversement du dipôle équivalent est une relaxation.Si maintenant on applique à l’échantillon une tension alternative V = V 0 exp (j 諸 t ), la relaxation ci-dessus se manifeste par une permittivité complexe équivalente, qu’on peut calculer exactement si V 0 est assez petit. Le résultat s’écrit:
où h est un paramètre qui caractérise l’aplatissement du diagramme.Pour h = 0, on retrouve la relation de Debye.Pour 0 諒 h 諒 1, le diagramme est bien un arc de cercle centré au-dessous de l’axe 﨎 , et la distribution f ( 精 ) correspondante a été calculée.Davidson et Cole ont effectué un travail analogue pour un diagramme en forme de demi-poire, incliné sur l’axe à 﨎 size=1秊, mais perpendiculaire à 﨎 s, tel qu’on l’observe souvent expérimentalement. Ces travaux sont fondamentaux, mais leur aspect macroscopique les rend peu exploitables.Depuis les développements de la mécanique statistique, et en particulier des travaux de R. Kubo, l’étude de la relaxation diélectrique en phase liquide a été entreprise à une échelle microscopique, et les travaux de S. H. Glarum ont ouvert une voie nouvelle vers l’interprétation des structures dipolaires en interaction. D’autre part, l’existence d’absorptions anormales a pu être attribuée aux effets d’inertie, qui peuvent devenir importants en haute fréquence. Notons enfin que des chercheurs de plusieurs universités françaises contribuent brillamment au développement de ces théories.10. Relaxation de charge d’espaceUn autre type important de relaxation est associé à l’accumulation de charges au voisinage d’électrodes plus ou moins bloquantes (c’est-à-dire plus ou moins impropres à décharger les ions arrivant à leur contact) sous l’influence combinée du champ, qui tend à accumuler les charges aux électrodes, et de l’agitation thermique qui, sous la forme du facteur de diffusion, s’oppose à cette accumulation.Considérons, par exemple, une plaque diélectrique d’un matériau de permittivité 﨎 contenant des charges libres entre électrodes bloquantes.Supposons que les charges des deux signes ont la même mobilité 猪 . Lorsqu’un champ permanent est appliqué entre les électrodes, les charges d’un même signe migrent vers l’électrode de signe opposé; cette migration est limitée par la diffusion, qui s’oppose à l’accumulation des charges. À l’état d’équilibre le matériau est macroscopiquement polarisé, et il contient une densité de charge 福 (x ), qui dépend de la distance x du point considéré aux électrodes. En d’autres termes, le matériau est équivalent à un dipôle (fig. 9). Si on inverse le sens du champ appliqué, la courbe de répartition des densités de charge évolue lentement vers la courbe b , symétrique de la courbe a par rapport à Ox, et, après un délai lié à la diffusion des charges, la configuration finale du matériau est équivalente à un dipôle opposé (fig. 9 b). Ce renversement du dipôle équivalent est une relaxation.Si maintenant on applique à l’échantillon une tension alternative V = V 0 exp (j 諸 t ), la relaxation ci-dessus se manifeste par une permittivité complexe équivalente, qu’on peut calculer exactement si V 0 est assez petit. Le résultat s’écrit: avec 﨡 = (d / D) 連1 + j 諸 精; D = 連D 精 (longueur de diffusion de Debye); 精 = 﨎 / 靖 (temps de relaxation de conduction):
avec 﨡 = (d / D) 連1 + j 諸 精; D = 連D 精 (longueur de diffusion de Debye); 精 = 﨎 / 靖 (temps de relaxation de conduction): La forme complexe de 﨎 ne permet pas une séparation simple des composantes 﨎 ( 諸 ) et 﨎 ( 諸 ), mais l’utilisation d’ordinateurs a permis de montrer que le diagramme d’Argand de la relation est très voisin d’un demi-cercle de Cole, mais avec un paramétrage en fréquence différent.11. Effet Maxwell-WagnerL’effet Maxwell-Wagner est la relaxation associée à un empilage de deux ou plusieurs feuilles de matériaux diélectriques de natures différentes.Les paramètres caractéristiques d’un empilage de deux feuilles et le circuit équivalent (par unité de surface) sont représentés sur la figure 10.Si la tension appliquée V est sinusoïdale, de pulsation 諸 , la permittivité équivalente du système est reliée à son admittance complexe face="EU Domacr" 喇 par unité de surface par la relation:
La forme complexe de 﨎 ne permet pas une séparation simple des composantes 﨎 ( 諸 ) et 﨎 ( 諸 ), mais l’utilisation d’ordinateurs a permis de montrer que le diagramme d’Argand de la relation est très voisin d’un demi-cercle de Cole, mais avec un paramétrage en fréquence différent.11. Effet Maxwell-WagnerL’effet Maxwell-Wagner est la relaxation associée à un empilage de deux ou plusieurs feuilles de matériaux diélectriques de natures différentes.Les paramètres caractéristiques d’un empilage de deux feuilles et le circuit équivalent (par unité de surface) sont représentés sur la figure 10.Si la tension appliquée V est sinusoïdale, de pulsation 諸 , la permittivité équivalente du système est reliée à son admittance complexe face="EU Domacr" 喇 par unité de surface par la relation: où C 0 est la capacité géométrique par unité de surface (C 0 = 﨎 0/d ). L’admittance complexe du système est ici:
où C 0 est la capacité géométrique par unité de surface (C 0 = 﨎 0/d ). L’admittance complexe du système est ici: avec:
avec: On peut mettre sous la forme:
On peut mettre sous la forme: avec:
avec: et:
et: Les deux premiers termes de (39) représentent une relaxation dipolaire du type de celle de Debye. Le terme additionnel imaginaire 1/j 諸 精0 introduit une déviation par rapport au demi-cercle de Cole et Cole (fig. 11).On peut montrer facilement que la différence ( s 漣 size=1秊) est proportionnelle à ( 精 1 漣 精 2)2. Il en résulte que si les matériaux 1 et 2 ont le même temps de relaxation diélectrique 精 1 = 精 2 = 精 , on a aussi s = r = = 精 / 精 0, de sorte que la relation (39) se réduit à:
Les deux premiers termes de (39) représentent une relaxation dipolaire du type de celle de Debye. Le terme additionnel imaginaire 1/j 諸 精0 introduit une déviation par rapport au demi-cercle de Cole et Cole (fig. 11).On peut montrer facilement que la différence ( s 漣 size=1秊) est proportionnelle à ( 精 1 漣 精 2)2. Il en résulte que si les matériaux 1 et 2 ont le même temps de relaxation diélectrique 精 1 = 精 2 = 精 , on a aussi s = r = = 精 / 精 0, de sorte que la relation (39) se réduit à: et le diagramme ( ) consiste en une demi-droite d’abscisse .Un autre cas particulier, d’une grande importance pratique, est celui où l’un des matériaux constituants est parfaitement isolant. Si, par exemple, 靖 1 est nul, on a 精 1 = 精 0 = 秊, et le troisième terme de (39) disparaît. Cette relation devient alors une relation de Debye, et le diagramme correspondant se réduit au demi-cercle de Cole et Cole, ce qui est d’ailleurs prévisible, puisque le circuit équivalent est, dans ce cas, un circuit de relaxation pure.La théorie de la relaxation de la polarisation interfaciale a été étendue par Wagner au cas d’une dispersion de sphérules d’un matériau dans un autre matériau, isolant et homogène. Le calcul montre qu’on a encore une relation de Debye, dont le temps de relaxation dépend, bien entendu, des permittivités des deux matériaux, et de la conductivité du matériau dispersé.La généralisation a même été poussée par Sillars au cas où les particules dispersées sont des sphéroïdes de révolution, et, dans ce cas, on a encore une relation de Debye, mais le temps de relaxation dépend aussi de l’excentricité des sphéroïdes.Ces résultats sont de nature macroscopique, en ce sens qu’ils admettent que la densité de charge électrique à l’intérieur des matériaux reste nulle.En fait, des mouvements de charges accompagnent forcément l’application d’un champ variable à un matériau composite. On peut s’en convaincre, par exemple, en notant que le potentiel, à l’interface médiane d’un empilement de deux matériaux soumis à un échelon de champ, passe de la valeur initiale imposée par la conservation de l’induction à l’interface, à la valeur finale imposée par la conservation du courant à l’interface.L’aspect microscopique lié au transfert et à l’accumulation des charges a été traité par Truckhan comme une généralisation du problème de l’évolution de la charge d’espace, et il a été discuté avec des hypothèses moins restrictives par Goffaux et Coelho.
et le diagramme ( ) consiste en une demi-droite d’abscisse .Un autre cas particulier, d’une grande importance pratique, est celui où l’un des matériaux constituants est parfaitement isolant. Si, par exemple, 靖 1 est nul, on a 精 1 = 精 0 = 秊, et le troisième terme de (39) disparaît. Cette relation devient alors une relation de Debye, et le diagramme correspondant se réduit au demi-cercle de Cole et Cole, ce qui est d’ailleurs prévisible, puisque le circuit équivalent est, dans ce cas, un circuit de relaxation pure.La théorie de la relaxation de la polarisation interfaciale a été étendue par Wagner au cas d’une dispersion de sphérules d’un matériau dans un autre matériau, isolant et homogène. Le calcul montre qu’on a encore une relation de Debye, dont le temps de relaxation dépend, bien entendu, des permittivités des deux matériaux, et de la conductivité du matériau dispersé.La généralisation a même été poussée par Sillars au cas où les particules dispersées sont des sphéroïdes de révolution, et, dans ce cas, on a encore une relation de Debye, mais le temps de relaxation dépend aussi de l’excentricité des sphéroïdes.Ces résultats sont de nature macroscopique, en ce sens qu’ils admettent que la densité de charge électrique à l’intérieur des matériaux reste nulle.En fait, des mouvements de charges accompagnent forcément l’application d’un champ variable à un matériau composite. On peut s’en convaincre, par exemple, en notant que le potentiel, à l’interface médiane d’un empilement de deux matériaux soumis à un échelon de champ, passe de la valeur initiale imposée par la conservation de l’induction à l’interface, à la valeur finale imposée par la conservation du courant à l’interface.L’aspect microscopique lié au transfert et à l’accumulation des charges a été traité par Truckhan comme une généralisation du problème de l’évolution de la charge d’espace, et il a été discuté avec des hypothèses moins restrictives par Goffaux et Coelho.
Encyclopédie Universelle. 2012.